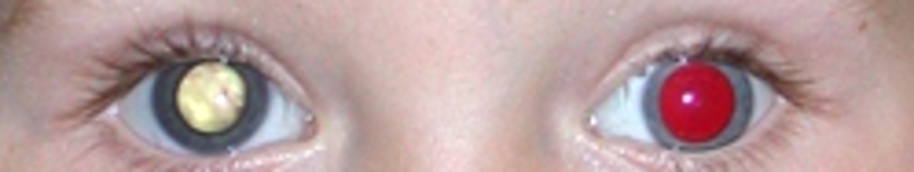
Was ist ein Retinoblastom?
Bei einem Retinoblastom spricht man von einem im Auge auftretendem Tumor,(intraokulare) der sich im Kindesalter mit einer Häufigkeit von einem betroffenen Kind auf etwa 18.000 Lebendgeburten
entwickeln kann. Diese Erkrankung ist anhand von Zahlen eher selten (in Deutschland ca. 60 Fälle/Jahr). Hierbei handelt sich um einen bösartigen (malignen) Tumor, der von genetisch veränderten
unreifen Netzhautzellen (Retinoblasten) ausgeht und unbehandelt zum Tod führt. Wird dieser jedoch frühzeitig erkannt und therapiert überleben mehr als 95 % der (meistens immer kleinen)
Patienten. Ist ein Auge betroffen, spricht man von einem unilateralen (einseitigen) Retinoblastom. Dies betrifft etwa 60% der erkrankten Kinder. Sind beide Augen erkrankt, handelt es sich
um ein bilaterales (beidseitiges) Retinoblastom.
Das Retinoblastom entwickelt sich in der Netzhaut und kann sich in verschiedene Richtungen ausbreiten. Bei einem Wachstum in Richtung des Glaskörpers (endophytisch) können sich Zellen von der
Oberfläche lösen und im Glaskörperraum schweben (Glaskörperaussaat des Tumors).
Erreichen diese Tumoranteile an anderen Stellen der Netzhaut, so entstehen intraokulare Absiedlungen, dadurch können mehrerer Tumore (multifokal) vorgetäuscht werden, obwohl ursprünglich ein
einzelner (solitären) Tumor vorlag. Diese Situation nennt man pseudomultifokales Wachstum. Infolge einer Glaskörperaussaat können auch Absiedlungen die Augenvorderkammer zwischen Hornhaut
(Cornea) und Regenbogenhaut (Iris) erreichen und das Bild einer inneren Entzündung des Auges imitieren, ähnlich einer bakteriellen Entzündung im Auge.
Findet ein Wachstum in Richtung Aderhaut (exophytisch) statt, kommt es oft zu einer Netzhautablösung. Ablösende Tumoranteile führen zu einer Absiedlung unter die Netzhaut (subretinal), somit besteht
die Gefahr für einen Einbruch (Infiltration) des Tumors in die Aderhaut. In sehr seltenen fortgeschrittenen Fällen kann der bösartige Tumor durch die Lederhaut (Sklera) wachsen und somit zu
einer Infiltration der Augenhöhle führen.
Was die Ausbreitung des Tumors entlang des Sehnervs betrifft, kann dies zu einer Mitbeteiligung des Gehirns, der Hirnhaut (Meningen) und des Hirnwassers (Liquor) führen. Durch einen Einbruch in
das Gefäßsystem kann es selten zu Tochtergeschwülsten (Metastasen) kommen, zum Beispiel ins Knochenmark, Lymphknoten, Knochen und Leber. Die regionalen Lymphknoten leiten die Lymphflüssigkeit
der vorderen Augenhöhle und der Haut ab. Sehr selten findet über diesen Weg eine Metastasierung statt. Das Retinoblastom kann grundsätzlich infolge eines kontinuierlichen Wachstums jede Struktur des
Auges infiltrieren.
Gefäßneubildungen in der Iris, das Absinken von in der Vorderkammer schwimmenden Tumorzellen, eine begleitende Entzündung der Augenhöhle (orbitale Zellulitis) oder ein erhöhter Augeninnendruck
(Sekundärglaukom) machen einen fortgeschrittenen Befund aus.
Das Retinoblastom tritt bei etwa 40% in beiden Augen auf (bilaterales RB). Es entsteht hierbei aber kein Tumorwachstum von einem Auge ins andere sondern mehrere Tumoren sind in beiden Augen entweder
zur gleichen Zeit aufgetreten oder können auch später noch wachsen. Somit kann sich ein einseitiges Retinoblastom zu einem späteren Zeitpunkt zu einem beidseitigem Retinoblastom entwickeln
(bilateral). Auch können sich in einem Auge mehrere, ganz unabhängig voneinander entstandene Tumoren entwickeln (multifokal).
Retinom
Diese Bezeichnung steht für ein spontan zurückgebildetes Retinoblastom, welches meist zufällig in 1-2 % bei der Untersuchung nicht erkrankter Verwandter von Retinoblastom erkrankten Kindern zu
finden ist. Es tritt jedoch nicht im Kindesalter auf und weist in aller Regel keine maligne Wachstumstendenz auf. Dieser Tumor erscheint wie ein Retinoblastom im inaktiven Rückbildungsstadium nach
Therapie. Auch dieser Befund sollte 1-2 mal pro Jahr untersucht werden, stellt aber in fast allen Fällen keine Bedrohung dar. Eine Behandlung ist hierbei nicht nötig. Auch ein Patient mit einem
Retinom sollte den Kontakt mit einem auf Retinoblastom spezialisierten Humangenetiker aufnehmen.
Trilaterales Retinoblastom
Bei einem trilateralem Retinoblastom handelt es sich um eine sehr seltene Kombination eines erblichen Retinoblastoms in einem oder beide Augen mit einem Tumor im Bereich der Mittellinie des Hirns
lokalisiert. Die in der Mittellinie liegende Zirbeldrüse (Glandula pinealis) ist am häufigsen betroffen. Der Zirbeldrüsentumor wird auch Pinealoblastom genannt. Grundsätzlich handelt es sich um
einen selbständig wachsenden Mittellinientumor und keine Metastase des Retinoblastoms aus den Augen. Es besteht eine histologische (feingewebliche) Ähnlichkeit zum Retinoblastoms. Die
Überlebensprognose ist gegenüber den Tumoren in den Augen deutlich schlechter.